





Abstract
Background. Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited human kidney disease and is caused by germline mutations in PKD1 (85%) or PKD2 (15%). It has been estimated that around 1% of tubular cells give rise to cysts, and cell hyperproliferation has been noted to be a cardinal feature of cystic epithelium. Nevertheless, it is uncertain whether the increase in proliferative index observed is an early or late feature of the cystic ADPKD kidney.
Methods. Two Pkd2 mouse mutants (WS25 and WS183) have been recently generated as orthologous models of PKD2. To determine the effect of Pkd2 dosage on cell proliferation, cyst formation and renal fibrosis, we studied renal tissue from Pkd2WS25/WS25 and Pkd2+/− mice by histological analysis. We also examined the proliferative index in archival nephrectomy tissue obtained from patients with ADPKD and normal controls.
Results. The proliferative index of non-cystic tubules in Pkd2 mutant mice as assessed by proliferating cell nuclear antigen and Ki67-positive nuclei was between 1–2%, values 5–10 times higher than control tissue. Similarly, the proliferative index of non-cystic tubules in human ADPKD kidneys was 40 times higher than corresponding controls. In Pkd2 mutant mice, significant correlations were found between the fibrosis score and the mean cyst area as well as with the proliferative index. Of significance, proliferating tubular cells were uniformly positive for polycystin-2 expression in Pkd2+/− kidney.
Conclusion. These results suggest that an increase in cell proliferation is an early event preceding cyst formation and can result from haploinsufficiency at Pkd2. The possible pathogenic link between tubular cell proliferation, interstitial fibrosis and cyst formation is discussed.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited renal disease and is caused by a spectrum of germline mutations in PKD1 (85%) or PKD2 (15%). The PKD1 protein, polycystin-1, is a large membrane glycoprotein thought to mediate cell–cell and/or cell–matrix interactions, whereas the PKD2 protein, polycystin-2, has homology to a family of voltage-gated cation channels (reviewed in [1]). Polycystin-1 interacts with polycystin-2 in vivo and the polycystin-1/polycystin-2 complex has been shown to function as a mechanosensory calcium channel in renal primary cilia [2] or as an adhesion complex mediating or regulating cell–cell or cell–matrix adhesion [3]. The disruption of this complex somehow leads to abnormalities in downstream cell signalling and results in the initiation of a cystic phenotype.
The mutational mechanism underlying cyst initiation in ADPKD remains controversial [4]. A two-hit mechanism of cyst formation has been proposed which could help explain the focal nature of cyst development and the variability of the cystic phenotype. Somatic mutations in the normal PKD1 or PKD2 allele are prerequisites for cyst initiation in this model. However, the continuing expression of polycystin-1 or polycystin-2 in the majority of ADPKD cysts [5] has suggested that cystogenesis may not always follow a simple ‘two-hit rule’. An alternative proposal is that haploinsufficiency itself may be sufficient to elicit a cystic or extrarenal non-cystic phenotype. Clinically, PKD2 hemizygosity in the deletion 4q21 syndrome is associated with a distinct phenotype including the early development of bilateral renal cysts [6]. Contiguous gene deletion of one allele each of PKD1 and the tuberous sclerosis-2 (TSC2) gene also causes a severe form of early-onset ADPKD [7].The strongest experimental evidence for haploinsufficiency has come from a recent study where a reduction in the level of Pkd1 was sufficient to initiate both renal and extrarenal (vascular) features of ADPKD [8].
While it is generally accepted that ADPKD cystic epithelial cells are hyperproliferative, it is not yet clear whether the increase in cell proliferation precedes cyst formation. Indeed, the observed increase in proliferative index (PI) of cyst lining cells noted in several studies of end-stage ADPKD renal tissue could imply that cell proliferation may be a late event, unrelated to cyst initiation but possibly a feature secondary to the process of cyst enlargement [9,10]. To re-evaluate the potential role of tubular cell proliferation in cyst initiation, we have studied the effect of haploinsufficiency of Pkd2 on cell proliferation, cyst formation and interstitial fibrosis in two mouse models with targeted deletions of Pkd2. We have also compared the proliferating activity in non-cystic renal tubules with cystic lining epithelia in human ADPKD tissue.
Subjects and methods
Animals and tissues
Two orthologous Pkd2 mouse models—an unstable allele (Pkd2WS25) and a true null allele (Pkd2WS183 or Pkd2−)—were generated as described previously [11,12]. The Pkd2WS25 allele comprises a mutant exon 1 introduced in tandem with the wild-type exon 1 at the mouse Pkd2 locus. It can undergo somatic recombination either to generate the wild-type allele (+) or a null allele (−) [11]. The Pkd2 mutant mice were backcrossed onto a C57BL/6 background for more than 10 generations. Colonies of both Pkd2 mutant mice were established from founder animals obtained from Prof. S. Somlo (Yale University). Eight Pkd2+/− (four male, four female) mice and four male Pkd2WS25/WS25 mice were bred and killed at 9–12 months of age. The mice had free access to tap water and standard mouse diet. Kidneys and liver were removed, fixed with 10% neutral buffered formalin for 24 h and paraffin embedded for histological examination. Nine aged-matched C57BL/6 mice were used as controls. All experimental procedures were carried out according to the rules and regulations laid down by the Home Office (Animal Scientific Procedures Act, UK 1986).
ADPKD patients and tissues
Stored archival kidney tissues (Department of Histopathology) were obtained from 16 patients (seven females and nine males) with ADPKD who had undergone nephrectomy for variable reasons, including intractable pain, huge abdominal masses, pre-transplantation, recurrent haematuria or cyst infection. All the tissues had been routinely formalin fixed and paraffin embedded. As normal controls, we used tissue obtained from the uninvolved pole of 10 kidneys removed because of a localized tumour. All archived materials (stored before AD 2000) were dissociated, and therefore, retrospective permission to use the tissue was not appropriate.
Histomorphometric analysis and fibrosis score
Haematoxylin/eosin-stained sections were used to determine the cyst number and mean cyst area. A cyst (cystically dilated tubule or Bowman's capsule) was defined as an abnormal epithelial lined cavity with dimensions greater than five times the normal tubular diameter. Transverse tissue sections (4 μm) with the largest cross-sectional area containing cortex, medulla and papilla of both kidneys were analysed. Renal fibrosis score was estimated by multiphase image analysis on Masson Trichrome-stained sections as the ratio of green staining of extracellular matrix to the red staining of cytoplasm. In each section, 10 randomized selected areas of cortex and medulla were analysed at a magnification of 200×. Image analysis was performed using the AnalySIS5™ software (Soft Imaging System GmbH, Germany).
Immunohistochemistry
Formalin-fixed paraffin sections were dewaxed and hydrated through graded ethanol solutions. For antigen retrieval, the sections were placed in 10 mM citric acid buffer (pH 6.0) and incubated in a pre-heated water-bath maintained at 95°C for 15 min for proliferating cell nuclear antigen (PCNA) staining, or boiled in a microwave oven for 25 min for Ki67 staining. Endogenous peroxidase activity was quenched by treatment with methanol containing 3% hydrogen peroxide for 30 min. Sections were then incubated in diluted blocking serum for 30 min, followed by incubation for 1 h in room temperature with a mouse anti-PCNA monoclonal antibody (PC10, DAKO) at 1 : 100 dilution or incubated overnight at 4°C with a rat anti-mouse Ki67 monoclonal antibody (TEC3, DAKO) at 1 : 25 dilution. A biotin-conjugated secondary antibody was used in human kidney sections followed by an avidin–biotin immunoperoxidase complex (ABC Elite, Vector Laboratories, Peterborough, UK), and developed using a commercial AEC kit (Sigma, UK). For mouse kidney sections, a peroxidase-conjugated secondary antibody was used and the peroxidase activity was demonstrated by a substrate solution of diaminobenzidine (DAB) containing 0.03% hydrogen peroxide. Negative control sections were processed with omission of the primary antibody. All sections were counterstained in haematoxylin.
Co-localization of polycystin-2 and PCNA
For double-labelling, tissue sections were incubated simultaneously with anti-PCNA and an antibody raised to the C-terminus of polycystin-2 (p30) overnight at 4°C [5]. The secondary antibodies used were: a peroxidase-conjugated goat anti-rabbit IgG (DAKO) for p30 staining and an alkaline phosphatase-conjugated goat anti-mouse IgG (DAKO) for PCNA staining. DAB and Vector Red (Vector labs, Peterborough, UK) were used as chromogens. The percentage of positive p30 cytoplasmic staining was counted in 100 PCNA-positive nuclei from renal sections from four Pkd2+/− mice.
Proliferative index
The PI was assessed by counting the percentage of PCNA or Ki67-positive renal tubules cells in five randomly selected fields (400×) of cortex and medulla per section. Between 500 and 1000 tubular epithelial nuclei were counted per section. Only clearly definable nuclei with heavy/or granular staining were identified as positive [9]. For human ADPKD renal sections, the PI of cyst-lining epithelia was also assessed in cysts with a non-attenuated cell lining.
Statistical analyses
Results are expressed as mean ± SEM. The statistical significance of differences between the groups was assessed with Kruskal–Wallis test, followed by a Dunn's multiple comparison test. Relationships between data were analysed using Spearman correlation. A P-value of <0.05 was considered statistically significant.
Results
Cyst formation in Pkd2 mutant mice
Overall, 75% of Pkd2WS25/WS25 and 62.5% of Pkd2+/− mice were found to have either renal and/or liver cysts. The severity of cystic disease was highly variable for Pkd2WS25/WS25 mice: one mouse had multiple liver and renal cysts (cyst number: 12; mean cyst area: 374 952 ± 157 663 μm2) (Figure 1A), another had a single large renal cyst (cyst area: 71 725 μm2), while a third had multiple liver cysts but no renal cysts. A fourth animal had no cysts (not shown). The cystic phenotype was similarly variable in Pkd2+/− mice: three had between 3–6 kidney cysts (mean cyst area: 87 257 ± 28 285 μm2) (Figure 1B), three had multiple liver cysts (Figure 1C), one of which also had kidney cysts but neither renal nor liver cysts were observed in three other mice.
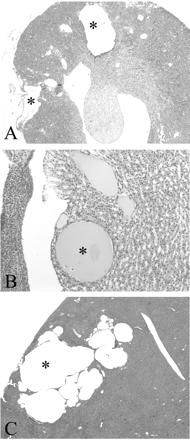
Histopathology of kidney and liver from Pkd2WS25/WS25 and Pkd2+/− mice. (A) Two large cortical cysts are shown in a Pkd2WS25/WS25 mouse with a severe cystic phenotype (20×). (B) A small renal cyst with flattened lining epithelium in the outer medulla of a Pkd2+/− mouse (400×). (C) Multiple liver cysts in a Pkd2+/− mouse (20×). *Indicates the lumen of larger cysts.
Proliferative index in Pkd2 mutant mice
The mean PI values of non-cystic tubules in Pkd2 mutant mice were 5–10 times higher than control mice as assessed using two cell proliferation markers, i.e. PCNA and Ki67 (PCNA PI: Pkd2±/−: 1.73 ± 0.38, Pkd2WS25/WS25: 1.08 ± 0.15, wild type: 0.19 ± 0.02; Ki67 PI: Pkd2±/−: 1.20 ± 0.19, Pkd2WS25/WS25: 1.09 ± 0.18, wild type: 0.17 ± 0.02). Statistical analysis indicated a highly significant difference between both Pkd2 mutant mice and wild-type control (Figure 2A). However, there was no significant difference between the PI of Pkd2WS25/WS25 and Pkd2+/− mice. PCNA-positive nuclei were located throughout the cortex and outer stripe of medulla with no clear predilection for nephron segments, frequently detected in the epithelial cells of normal height and appearance (Figure 2B and C). Clusters of PCNA-positive cells were occasionally observed, mainly in areas surrounding a neighbouring cyst (Figure 2D). Most cysts were lined by flattened epithelial cells with only an occasional cyst lined by cuboidal epithelium. An example of a cyst-lining cell displaying PCNA positivity is shown in Figure 2E. However, we could not accurately estimate the PI for cyst-lining cells in Pkd2 mouse kidney because of the scarcity of this type of cyst. Occasional proliferating cells could also be observed in the interstitium near PCNA-positive tubular cells (Figure 2F). Although the PI with Ki67 was slightly lower than that measured with PCNA, the expression pattern of Ki67 staining between the three genotypes was similar to that of PCNA (Figure 2G–I).
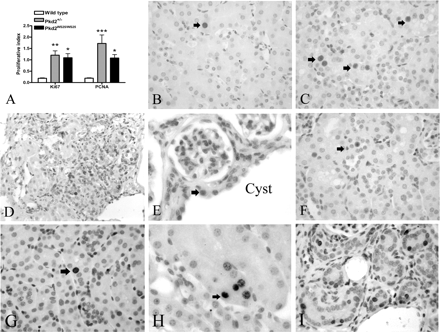
Tubular cell proliferation in Pkd2 mutant mice measured by PCNA and Ki67 immunostaining. (A) The PI in non-cystic tubules of wild-type (n = 9), Pkd2WS25/WS25 (n = 4) and Pkd2+/− (n = 8) kidneys. PI values were 5–10 times higher in both Pkd2 mutant kidneys compared with wild-type (*P < 0.05, **P < 0.01, ***P < 0.001). (B) Few positive PCNA-staining nuclei (arrow) were detected in wild-type kidney (400×). (C) Multiple PCNA-positive nuclei (arrows) could be found in normal tubular cells in Pkd2+/− kidney (400×). (D) Occasional clusters of PCNA-positive nuclei were found in some tubules adjacent to a larger cyst in Pkd2WS25/WS25 kidney (200×). (E) A single PCNA-positive cuboidal cyst-lining cell (arrow) from Pkd2WS25/WS25 kidney (600×). (F) A PCNA-positive cell (arrow) in the interstitium between several positive PCNA-staining tubular cells (400×). (G) Occasional positive Ki67-staining nuclei (arrow) in wild-type kidney (400×). (H) Several Ki67-positive cells detected in a non-cystic tubule (arrow) in Pkd2+/− kidney (600×). (I) Multiple Ki67-positive cells found in non-cystic tubules and interstitium in Pkd2WS25/WS25 kidney (400×).
Proliferative index in human ADPKD kidney
In ADPKD, the PI of tubular epithelium was markedly increased in both non-cystic renal tubules and cystic lining epithelia (Figure 3A). The PI values of non-cystic tubules were elevated by 40-fold compared with controls (1.19 ± 0.15 vs 0.03 ± 0.02, P < 0.001) (Figure 3A). The PI value of cyst epithelial lining was around 18-fold higher than that of controls (0.54 ± 0.13 vs 0.03 ± 0.02, P < 0.01), but not significantly different from that of non-cystic ADPKD tubules. The average age of the control cases was not different from ADPKD patients (59.7 ± 4.6 vs 50.4 ± 1.7 years, P > 0.05). In some sections, severe fibrosis and inflammatory cells were frequently seen in addition to cysts at different stages and dilated tubules. PCNA-positive cells were detected in tubules of normal appearance as well as in all types of cysts having different thicknesses of renal epithelium (Figure 3B and C). However, larger cysts tended to have fewer PCNA-positive cells (data not shown). Some PCNA-positive cells were seen in the interstitium adjacent to PCNA-positive renal tubules or cysts (Figure 3D).
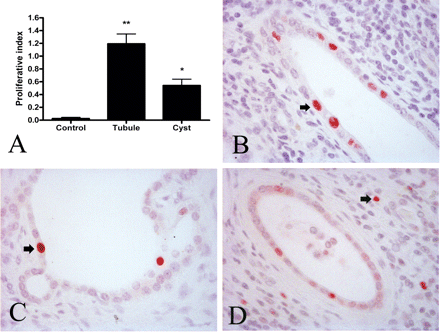
Tubular and cystic cell proliferation in human normal and ADPKD kidney. (A) The PI of control kidneys (n = 10) compared with ADPKD kidneys (n = 16). The PI for cystic kidney was calculated separately for non-cystic tubules and cysts and each was significantly different compared with control values (*P < 0.01, **P < 0.001). Examples of PCNA-positive nuclei (arrows) in a normal tubule (B), a cyst (C) and interstitium (D). Magnification: 400×.
Fibrosis score
Masson Trichrome staining in kidney sections from both Pkd2 mutant mice revealed increased fibrosis especially in areas surrounding renal cysts (Figure 4A–C). The fibrosis scores of Pkd2+/− (0.0204 ± 0.0069) and Pkd2WS25/WS25 (0.0388 ± 0.0258) kidneys were significantly higher than those of normal controls (0.0006 ± 0.0004, P < 0.05) but were not significantly different from each other.
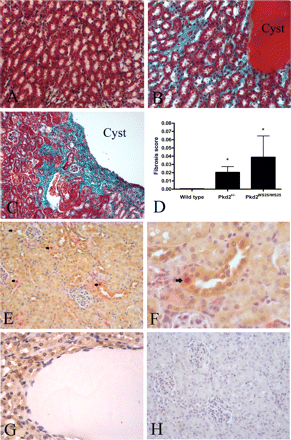
Renal fibrosis in Pkd2 mutant mice (A–D) and localization of PCNA and polycystin-2 in Pkd2+/− kidneys by immunohistochemistry (E–H). Masson Trichrome staining was used to demonstrate the presence of interstitial collagen (green). Cell cytoplasm stains red. (A) Wild-type control kidney showed little fibrosis. (B) Increased renal fibrosis can be seen surrounding a medullary cyst in Pkd2+/− kidney. (C) Severe fibrosis surrounding a large cortical cyst in Pkd2WS25/WS25 kidney. (D) Fibrosis scores for Pkd2+/− (0.0204 ± 0.0069) and Pkd2WS25/WS25 (0.0388 ± 0.0258) kidneys were significantly higher than wild-type controls (0.0006 ± 0.0004) (*P < 0.05 vs control). Magnification: 200×. (E) In Pkd2+/− and normal (not shown) kidneys, polycystin-2 expression (brown) was strongest in the distal convoluted tubules and cortical collecting duct. Several nuclei staining positive for PCNA (red, arrowheads) can also be seen positive for polycystin-2 (brown) (200×). (F) Higher power view of a PCNA-positive cell in a distal tubule showing cytoplasmic polycystin-2 staining (arrow) (400×). (G) An example of a cyst with cyst-lining negative for polycystin-2 staining (400×). (H) No detectable expression in a negative control section (200×).
Co-localization of polycystin-2 and PCNA
The expression of polycystin-2 in Pkd2+/− kidneys was examined by immunohistochemistry (Figure 4E–H). As previously described in normal human [5] and rat [13] kidney, polycystin-2 expression was more prominent in the distal convoluted tubules and cortical collecting ducts, weaker in proximal tubules and not seen in glomeruli. Most PCNA-positive cells were also positive for polycystin-2 (Figure 4F). Among the 100 PCNA-positive nuclei counted in renal sections from four Pkd2+/− mice, 94 cells were also clearly positive for polycystin-2. Most cyst-lining epithelial cells were negative for polycystin-2 staining (Figure 4G). No staining was seen in negative control sections (Figure 4H).
Correlation between scores for renal cysts, fibrosis score and PI
We further examined the relationships between renal cyst number, mean cyst area, fibrosis score and the PI of non-cystic tubules in Pkd2 mutant mice. Fibrosis score correlated significantly with the mean cyst area (R = 0.79, P = 0.0002) (Figure 5A) as well as the number of renal cysts (R = 0.78, P < 0.002) (Figure 5B). The fibrosis score also correlated with PI (PCNA PI: R = 0.65, P = 0.0047; Ki67 PI: R = 0.79, P = 0.0003)(Figure 5C). There was, however, no significant correlation between PI and the number of renal cysts (data not shown) or the mean cyst area (PCNA PI: R = −0.19, P = 0.56; Ki67 PI: R = 0.48, P = 0.20) (Figure 5D).
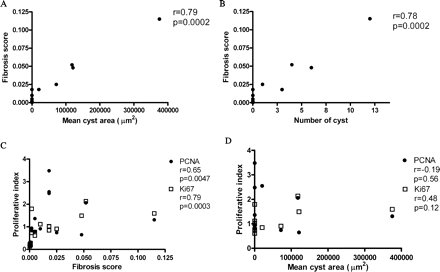
Correlations between cyst number, fibrosis score and PI. (A) Fibrosis score correlated with mean cyst area (R = 0.79, P = 0.0002). (B) Fibrosis score correlated with the number of renal cysts (R = 0.78, P = 0.0002). (C) Fibrosis score also correlated with PCNA PI (R = 0.65, P = 0.0047) and Ki67 PI (R = 0.79, P = 0.0003). (D) There was no significant correlation between PI and mean cyst area (PCNA: R = −0.19, P = 0.56; Ki67: R = 0.48, P = 0.12).
Discussion
The major finding of this study was an unexpected increase in the PI in the normal (non-cystic) tubules of Pkd2+/− and Pkd2WS25/WS25 mice. A similar increase was observed in the normal tubules of end-stage ADPKD human kidney, although the genotypes of these patients were not known. These results suggest that increased tubular cell proliferation is likely to precede cyst formation in ADPKD. Previous studies have reported a 10–100-fold increase in the PI of non-cystic tubules in end-stage human ADPKD kidney as determined by Ki67 or PCNA staining [9,10]. An increase in cell proliferation appears to be an early event preceding cystic dilatation of the nephron in the Han:SPRD rat as well as in the early stage of cyst growth in compound heterozygous Pkd2WS25/− mouse [14–16]. In these studies, proliferative activity was present not only in the epithelial cells of cysts but also in the non-dilated tubules and interstitial cells.
We confirmed previous findings that polycystin-2 expression is absent in the renal cysts of both Pkd2 mutant mice strains [11,17]. However, we noted that the vast majority of PCNA-positive cells (94%) remained polycystin-2 positive regardless of their origin. This finding suggests that the increase of proliferation in renal tubular epithelium cannot be attributed simply to somatic mutation of the normal Pkd2 allele in individual cells. An alternative explanation for the increase in PI could be haploinsufficiency of Pkd2. Our data are consistent with previous studies suggesting that haploinsufficiency at Pkd2 can induce phenotypic changes in non-renal cells [18] and provide evidence that Pkd2 haploinsufficiency also influences the proliferative activity of renal tubular epithelium.
No significant correlation between PI and cyst number or mean cyst area was observed in this study. In addition, the PI in human ADPKD cysts was lower than that in non-cystic tubules (Figure 2). One possible explanation is that tubular cell proliferation is an early event in the birth of cysts but does not persist as cysts enlarge (Figure 6) [14,15,19]. These results could indicate that an increase in proliferative activity alone may be insufficient for cysts to progress. Significantly, not all Pkd2+/− mice developed renal (or liver) cysts despite the increase in PI, so it seems likely that other factors, such as environmental factors, stochastic events and modifying genes additionally modify cyst growth.
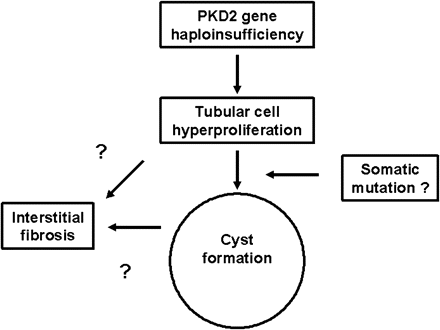
Hypothetical scheme of the relationships between PKD2 haploinsufficiency, cell proliferation, cyst formation and interstitial fibrosis. In this scheme, the increase in cell proliferation may predispose the tubular cell to a somatic mutation, necessary for cystic change. The significant correlations between PI and mean cyst area with the fibrosis score (Figure 5) suggest that interstitial fibrosis could potentially result directly from the secretion of tubular-derived cytokines or extracellular matrix proteins in two phases, i.e. early from proliferating cells in non-cystic tubules, or late from cyst epithelial cells.
The diseased ADPKD kidney is not only characterized by the formation of cysts but is also associated with changes in extracellular matrix and interstitial inflammation which if progressive, result in fibrosis and loss of renal function [1]. In the present study, we observed significant correlations between the fibrosis score and mean cyst area as well as the PI. The former could reflect a compensatory increase in collagen deposition in regions adjacent to larger cysts but the latter could imply that tubular cell proliferation could directly lead to interstitial fibrosis. In this context, it is interesting to note that rapamycin has been shown to slow disease progression in the Han:SPRD rat while simultaneously reducing the number of PCNA-positive cells in non-cystic tubules [20]. Although speculative, this association merits further investigation.
In conclusion, our results suggest that haploinsufficiency at Pkd2 alone can result in increased tubular proliferation and interstitial fibrosis in the cystic kidney. Since the increase in cell proliferation was prominent in non-cystic tubules, it probably represents one of the earliest events in cyst formation. The correlation between PI and fibrosis score suggests that therapeutic strategies to reduce cell proliferation could potentially influence not only cyst formation but also the process of renal scarring in ADPKD kidney.
This work was supported by grants from the Polycystic Kidney Disease Foundation and the Special Trustees of the Sheffield Teaching Hospitals, UK. M.Y.C. was supported by a Biomedical Scholarship from Chang Gung Memorial Hospital, Taiwan. We thank Stefan Somlo for helpful comments on the manuscript and Salwa Ismail for technical assistance.
Conflict of interest statement. None declared.
References
Ong AC, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex.
Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells.
Streets AJ, Newby LJ, O’Hare MJ, et al. Functional analysis of PKD1 transgenic lines reveals a direct role for polycystin-1 in mediating cell-cell adhesion.
Ong AC, Harris PC. Molecular basis of renal cyst formation – one hit or two?
Ong AC, Ward CJ, Butler RJ, et al. Coordinate expression of the autosomal dominant polycystic kidney disease proteins, polycystin-2 and polycystin-1, in normal and cystic tissue.
Velinov M, Kupferman J, Gu H, et al. Polycystic kidneys and del (4)(q21.1q21.3): further delineation of a distinct phenotype.
Brook-Carter PT, Peral B, Ward CJ, et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease – a contiguous gene syndrome.
Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease.
Nadasdy T, Laszik Z, Lajoie G, Blick KE, Wheeler DE, Silva FG. Proliferative activity of cyst epithelium in human renal cystic diseases.
Lanoix J, D’Agati V, Szabolcs M, Trudel M. Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD).
Wu G, D’Agati V, Cai Y, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease.
Wu G, Markowitz GS, Li L, et al. Cardiac defects and renal failure in mice with targeted mutations in Pkd2.
Zhao Y, Haylor JL, Ong AC. Polycystin-2 expression is increased following experimental ischaemic renal injury.
Ramasubbu K, Gretz N, Bachmann S. Increased epithelial cell proliferation and abnormal extracellular matrix in rat polycystic kidney disease.
Thomson RB, Mentone S, Kim R, et al. Histopathological analysis of renal cystic epithelia in the Pkd2WS25/− mouse model of ADPKD.
Kuehn EW, Park KM, Somlo S, Bonventre JV. Kidney injury molecule-1 expression in murine polycystic kidney disease.
Wu G, Tian X, Nishimura S, et al. Trans-heterozygous Pkd1 and Pkd2 mutations modify expression of polycystic kidney disease.
Qian Q, Hunter LW, Li M, et al. Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells.
Nishio S, Hatano M, Nagata M, et al. Pkd1 regulates immortalized proliferation of renal tubular epithelial cells through p53 induction and JNK activation.
Author notes
1Academic Nephrology Unit, Sheffield Kidney Institute, Division of Clinical Sciences (North), University of Sheffield, Sheffield, 2Department of Histopathology, Sheffield Teaching Hospitals Foundation Trust, Northern General Campus, Sheffield, UK and 3Department of Medicine, Cairo University, Egypt


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments