





Abstract
Elevated plasma levels of the phosphaturic hormone fibroblast growth factor 23 (FGF-23) are a hallmark of chronic kidney disease (CKD)-mineral and bone disorder. FGF-23 allows serum phosphate levels within physiological limits to be maintained in progressive CKD until end-stage renal disease is reached. Despite its seemingly beneficial role in phosphate homeostasis, several prospective studies in dialysis patients and in patients with less advanced CKD associated elevated FGF-23 with poor cardiovascular and renal outcome. Moreover, very recent evidence suggests an adverse prognostic impact of elevated FGF-23 even in subjects without manifest CKD. These epidemiological data are supplemented by laboratory findings that reveal a pathophysiological role of FGF-23 in the pathogenesis of myocardial injury. In aggregate, these clinical and experimental data identify FGF-23 as a promising target of novel therapeutic interventions in CKD and beyond, which should be tested in future clinical trials.
Introduction
Patients with chronic kidney disease (CKD) suffer from tremendous cardiovascular morbidity and mortality. This is exemplarily illustrated by recent data from the ERA-EDTA Registry which found 35-year-old incident dialysis patients to share the cardiovascular prognosis with 75-year-old subjects from the general population [1]. Numerous clinical cohort studies published in the last decade indicate that cardiovascular morbidity may even increase in much earlier CKD stages, as summarized by the Chronic Kidney Disease Prognosis Consortium [2].
It is generally acknowledged that this high cardiovascular risk in CKD patients is not merely caused by ‘traditional’ risk factors that drive vascular disease in the general population—namely smoking, hyperglycaemia, hypercholesterolaemia, arterial hypertension, obesity and physical inactivity [3]. In line with this assumption, prediction models derived from the general population do not adequately predict future cardiovascular events among CKD patients [4]. Instead, CKD-specific risk factors undoubtedly contribute to cardiovascular morbidity in these patients [3].
This concept is corroborated by the failure of most interventional studies which aimed to target traditional cardiovascular risk factors in CKD [5–7]. Thus, a clinical update from Kidney Disease: Improving Global Outcome advocated to clarify the role of novel risk factors as potential new therapeutic targets [3].
A considerable number of CKD-specific risk factors have been defined in recent years. Unfortunately, some of these factors are hard to tackle therapeutically—such as chronic microinflammation—while others have been targets of recent trials which uniformly yielded disappointing results: correction of anaemia [8], vitamin substitution in hyperhomocysteinaemia [9–11], early dialysis initiation [12], intensification of dialysis treatment or the use of modern dialysis equipment [13] failed to improve prognosis of CKD patients.
Against this background, CKD-mineral and bone disorder (CKD-MBD) represent a welcome exception: first, several components of CKD-MBD have consistently been associated with adverse outcome in epidemiological studies [14–17]. Secondly, therapeutic agents for CKD-MBD are widely available. Thirdly, cohort studies [18] and preliminary interventional trials on surrogate markers [19–21] suggest that modification of the CKD-MBD parameters may be beneficial, although definite data from adequately powered studies analysing ‘hard’ cardiovascular endpoints are pending.
Our conceptual understanding of CKD-MBD has been challenged by the discovery of the phosphaturic hormone fibroblast growth factor 23 (FGF-23). Only 12 years after its first description, most experts agree that FGF-23 plays a central role in CKD-MBD, and very recent data suggest a direct pathophysiological link between FGF-23 and cardiovascular disease in CKD.
In the present paper, we will first give a short summary of the physiological role of FGF-23 in human calcium–phosphate metabolism. Several detailed reviews on this topic have been published in the last years, to which the interested reader may refer [22–25]. Next, we will discuss the tremendous rise in FGF-23 levels (for which the term ‘hyperphosphatoninism’ was coined) in CKD patients, and we will review epidemiological studies which associated hyperphosphatoninism with prevalent and incident cardiovascular disease. Finally, we will summarize models which offer pathophysiological explanations for these associations, and discuss FGF-23 as a future therapeutic target in CKD.
Physiological functions of FGF-23
FGF-23 is a 251-amino acid protein with a molecular weight of 26 000 Da, which is mainly synthesized and secreted by osteoblasts [26]. Together with FGF-19 and FGF-21, FGF-23 belongs to the FGF-19 subfamily of FGFs. Unlike most other FGFs, which bind to heparin sulphate in the extracellular matrix and thus exert paracrine and autocrine effects, members of the FGF-19 subfamily display topological differences in the heparin-binding region. Consequently, FGF-23 is less avidly captured in the extracellular matrix, and thus more prone to enter the systemic circulation, allowing FGF-23 to exert endocrine functions [27, 28].
It is generally assumed that hyperphosphataemia and/or oral phosphate loading induces osteoblastic FGF-23 secretion, although the exact mechanisms remain enigmatic at present. Calcitriol and parathyroid hormone (PTH) are additional regulators of FGF-23 secretion, as discussed below.
FGF-23 exerts its physiological functions by binding to FGF receptors (FGF-R) on the surface of target cells in the presence of membrane-bound Klotho, which serves as a co-receptor. Of note, FGF-Rs are found ubiquitously. In contrast, Klotho is selectively expressed in kidneys, parathyroid glands and in the choroid plexus [29], rendering these organs physiological targets for FGF-23.
In the kidney, FGF-23 mediates its phosphaturic effect by reducing the expression and activity of sodium–phosphate (Na/Pi) cotransporters in the proximal tubules [30]. Additionally, FGF-23 may reduce intestinal NaPi cotransporter activity and thus directly inhibit intestinal phosphate absorption [31].
Furthermore, FGF-23 lowers serum calcitriol levels by decreasing the renal expression of 1α-hydroxylase (CYP27B1), which is the rate-limiting step in calcitriol synthesis, and increasing the expression of 24-hydroxylase (CYP24A1), which degrades calcitriol [32]. As calcitriol itself stimulates FGF-23 generation by binding to the FGF-23 gene promoter [33], a regulatory feedback loop between FGF-23 and calcitriol exists.
Finally, FGF-23 suppresses PTH synthesis in vitro [34] and in vivo [35]. However, parathyroid glands may become resistant towards FGF-23 in uraemia, as PTH levels are elevated in the presence of severe hyperphosphatoninism in uraemic rodent models [36].
In contrast, it is uncertain whether PTH directly affects FGF-23 secretion: no effect of PTH on FGF-23 expression in bone cells was found in vitro [33, 37, 38] and conflicting in vivo data on the impact of PTH on FGF-23 levels have been reported from animal studies [37, 39] and in clinical trials [40–42]. Physiological interactions may partly explain these contradictory data, as PTH directly modifies phosphate and calcitriol levels, which themselves affect FGF-23 secretion. It has been suggested that a biphasic effect may exist, with an initial decrease in FGF-23 levels after PTH application to counteract PTH-induced phosphaturia, and a later increase in FGF-23 [41]. In summary, the exact regulatory circuit between FGF-23 and PTH is not fully understood yet. The central role of FGF-23 in phosphate metabolism is summarized in Figure 1.
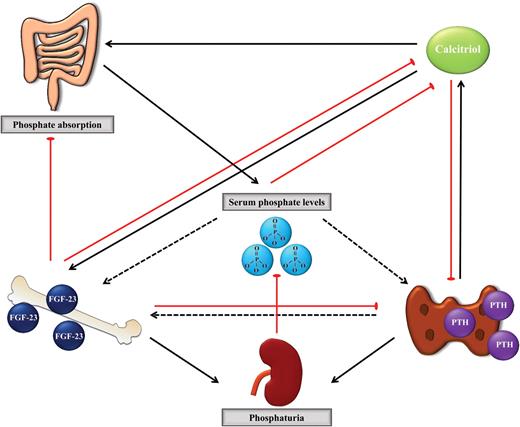
Serum phosphate regulation. Black arrows indicate activating pathways; red lines indicate inhibitory pathway. Note the exact pathways by which phosphate triggers PTH and FGF-23 secretion, and the direct link between PTH and FGF-23 await further experimental studies, as indicated by dotted arrows. Details are discussed in the manuscript.
Measurement of FGF-23
Two assays for quantification of human FGF-23 are commercially available. Full-length (‘intact’) FGF-23 levels can be determined with a sandwich enzyme-linked immunosorbent assay, in which two different monoclonal antibodies detect the simultaneous presence of both the N-terminal and C-terminal portions of FGF-23. In contrast, the ‘C-terminal’ assay recognizes both full-length and processed C-terminal fragments of FGF-23. Measurement results from the ‘intact’ and the ‘C-terminal’ assay are strongly correlated, and most experts agree that the use of either of these assays is adequate. For the ease of legibility, the present review will not indicate the specific assay used in individual clinical studies.
FGF-23 in CKD
A progressive rise in FGF-23 occurs over the spectrum of CKD [43, 44], and maintenance dialysis patients may display FGF-23 levels which are more than 100-fold higher compared with healthy subjects [43]. Thereby, FGF-23 levels of most dialysis patients by far surpass levels that are observed in patients with hereditary diseases of phosphate metabolism, such as hypophosphataemic rickets [45].
Recent papers expanded these findings and reported an increment in FGF-23 levels already at very early stages of CKD [46]. More than half of all CRIC (Chronic Renal Insufficiency Cohort Study) participants with slightly reduced kidney function [defined as estimated glomerular filtration rate (eGFR) 50–59 mL/min] had elevated FGF-23 levels, while PTH was elevated only in a minority of these patients, and virtually, no patient had hyperphosphataemia [47]. Elevated FGF-23 is therefore considered as the earliest detectable change in CKD-MBD.
Still, it remains unclear which stimulus drives this increase in FGF-23 in the early course of CKD. Two opposing hypotheses were raised: either kidney injury itself may induce the production of a—yet to be defined—factor that stimulates FGF-23 section (or alternatively, decreased the production of a tonic inhibitor) [47]. This hypothesis is partly supported by animal data that demonstrate an early increase in FGF-23 after kidney injury [48]. Alternatively, increased levels of FGF-23 may reflect dampened Klotho expression in target organs [49, 50]. In line with this hypothesis, decreased renal Klotho expression has been detected in kidney biopsy studies [51]. As FGF-23 requires Klotho for its phosphate-regulating effects in kidneys and in parathyroid glands, higher FGF-23 might physiologically be needed to overcome Klotho deficiency and to maintain normophosphataemia in states of low Klotho expression.
If this second hypothesis held true, hyperphosphataemia and hypervitaminosis D should at least transiently occur in early CKD as a reflection of such initial Klotho deficiency [47]. Of note, large epidemiological data stand against this hypothesis, as low rather than high serum phosphate values are found in very early CKD [47]. As FGF-23 itself down-regulates Klotho expression [52], Klotho deficiency in early CKD might thus reflect a primary increase in FGF-23.
Implications of hyperphosphatoninism in CKD
Counterintuitive to the presumptively protective, phosphate-regulating properties of FGF-23, several longitudinal studies associated hyperphosphatoninism with adverse renal outcome and mortality in CKD patients.
First prospective data came from the Mild to Moderate Kidney Disease (MMKD) study, which investigated the relationship between FGF-23 and CKD progression in 177 non-diabetic CKD stage 1–5 patients not receiving dialysis therapy [53]. Over a median follow-up of 4.4 years, patients with elevated FGF-23 (defined as C-terminal FGF-23 >104 rU/mL and/or intact FGF-23 >35 pg/mL) more often reached the primary combined endpoint defined as doubling of serum creatinine and/or onset of renal replacement therapy. Given the strong association between FGF-23 and renal function at the baseline, such a correlation may be expected in univariate analysis. However, this association remained significant after adjustment for confounders including eGFR. Similar findings were reported in a smaller cohort of 55 diabetic CKD patients prospectively followed for 31 months [54].
Subsequently, the association between FGF-23 and renal outcome was re-analysed in two large longitudinal cohort studies, namely CRIC [55] (3879 CKD stage 2–4 patients; median follow-up 3.5 years) and HOST [56] (Homocysteine in Kidney and End-Stage Renal Disease study; 1099 CKD stage 4–5 patients not on dialysis; median follow-up 2.9 years), which yielded partly conflicting results. Both studies assessed renal outcome (defined as initiation of dialysis treatment or renal transplantation in CRIC, and as initiation of dialysis treatment in HOST) and patient survival; HOST additionally reported cardiovascular events. In line with MMKD, both studies found high FGF-23 as a predictor of adverse renal outcome in univariate analyses. This relationship lost significance after adjusting for eGFR in the CRIC study, but not in HOST. In subgroup analyses, elevated FGF-23 levels remained an independent renal predictor in CRIC participants with a baseline eGFR of >30 mL/min/1.73 m2, but not in patients with more severe CKD at the baseline [55].
Nonetheless, CRIC and HOST consistently found an increasing mortality risk at higher quartiles of FGF-23. Compared with the lowest FGF-23 quartile, patients in the fourth quartile suffered from a more than 2-fold higher mortality after adjustment for classical cardiovascular risk factors and traditional CKD-MBD markers [55, 56].
These findings are congruent with earlier data from cohort studies in CKD 5D patients: in a nested case–control analysis within the Accelerated Mortality on Renal Replacement (ArMORR) study, Gutierrez et al. [57] measured FGF-23 levels of 400 incident haemodialysis patients. Elevated FGF-23 levels not only independently predicted 1-year all-cause mortality, but even surpassed serum phosphate levels as a marker of adverse outcome [45, 57]. Similar findings were reported in a cohort of 219 prevalent dialysis patients followed for 2 years [58]; notably, a third dialysis study reported FGF-23 to predict survival only among male patients with prevalent cardiovascular disease [59].
FGF-23 and cardiovascular outcome
Unfortunately, none of these CKD studies that associated elevated FGF-23 with higher mortality reported specific causes of death [55–59], and only HOST prospectively analysed cardiovascular events [56]. Here, patients in the highest quartile of baseline FGF-23 had a hazard ratio of 2.58 for future cardiovascular events in univariate analysis, compared with patients with lowest FGF-23. In multivariate analysis, high FGF-23 remained a significant predictor of cardiovascular outcome, while native vitamin D, calcitriol and PTH did not. When separating the composite cardiovascular endpoint into its individual components, elevated FGF-23 predicted myocardial infarction and lower extremity amputation, but not thrombotic stroke [56].
These findings are in agreement with an earlier analysis from our group among 149 CKD stage 1–5 non-dialysis patients, in which elevated FGF-23 independently predicted a predefined combined cardiovascular endpoint. Our study was not powered to separately analyse mortality or individual components of the combined cardiovascular endpoint [60]. Finally, the Osaka Vitamin D Study in Patients with CKD (OVIDS-CKD) very recently aimed to extend these findings to Asian patients. OVIDS-CKD recruited 738 CKD stage 1–5 patients not on dialysis from Japan and again found high FGF-23 to predict broadly defined cardiovascular events when censoring patients at initiation of chronic dialysis treatment. This finding was less evident when further following patients after the onset of dialysis treatment, and analysis of cause-specific mortality was hampered by the low cardiovascular death toll [61].
In summary, across the stages of CKD, FGF-23 may predict all-cause mortality, cardiovascular event rate and renal progression. These findings inspired several groups to inquire whether a similar prognostic role of FGF-23 may exist in subjects without overt CKD. Such an idea follows recent reports which identified other components of CKD-MBD (e.g. serum phosphate [62–65] and hypovitaminosis D [66]) as predictors of adverse outcome in subjects without overt kidney disease.
Presently, longitudinal data from three cohort studies [namely the Heart and Soul Study [67], Health Professionals Follow-up Study (HPFS) [68] and the Women's Health and Aging Study (WHAS) [69]] have been published. Results from the Prevention of Events with Angiotensin Converting Enzyme Inhibition (PEACE) trial were presented at the American College of Cardiology's 61st Annual Scientific Session [70], and data from our HOM SWEET HOMe study will be submitted for presentation at the American Society of Nephrology's kidney week in late 2012.
In the Heart and Soul Study, FGF-23 was measured in 833 outpatients with prevalent coronary heart disease, who were followed for a median of 6 years. High FGF-23 at the baseline independently predicted adverse outcome. After adjustment for confounders, risk for incident cardiovascular events and mortality was doubled in the highest FGF-23 tertile compared with the lowest tertile [67]. A nested case–control analysis from HPFS yielded seemingly conflicting results: baseline FGF-23 levels did not differ between 422 study participants who subsequently suffered a coronary event and 837 age-matched controls who had no event over a follow-up period of 10 years [68].
Next, the WHAS study analysed FGF-23 in 701 moderately to severely disabled women (half of whom had impaired eGFR at baseline), who were followed for a median of 18 months. Among those women with eGFR ≥60 mL/min/1.73 m2 at the baseline, higher baseline FGF-23 was associated with incident CKD during follow-up. In contrast, FGF-23 did not independently predict global or cause-specific mortality. Of note, only 18.6% deaths were attributed to cardiovascular disease, which may result from selective inclusion of disabled women. Short follow-up, missing data on non-fatal cardiovascular events and high prevalence of CKD at the baseline are further limitations, which in aggregate preclude an uncritical transfer of these results from highly selected disabled seniors to subjects from the general population.
Instead, the discrepant results from the Heart and Soul Study [67] and from HPFS [68] warrant a critical review of major differences in study design. First, the Heart and Soul Study recruited patients with prevalent coronary artery disease, whereas HPFS included subjects without cardiovascular disease at the baseline. Secondly, different endpoints were assessed, as HPFS focused on coronary events (non-fatal acute myocardial infarction and fatal coronary heart disease), whereas Heart and Soul analysed a combined endpoint which comprised acute myocardial infarction and several non-coronary events (namely stroke, transient ischaemic attacks and admission for heart failure). Interestingly, the Heart and Soul Study investigators reported the impact of FGF-23 measurements on individual components of the combined endpoints separately; elevated FGF-23 selectively predicted non-coronary events, particularly admission for heart failure, while it failed to predict acute myocardial infarction—quite in line with HPFS. These findings are supplemented by very recently presented data from the PEACE trial [70]: PEACE randomized 8290 patients with stable coronary artery disease and normal or near-normal left ventricular function to intake of the ACE inhibitor trandolapril or placebo. The PEACE trial investigators now performed a nested case–control study which comprised 1100 out of 8290 PEACE participants, in whom FGF-23 and traditional cardiovascular biomarkers were analysed from samples drawn at study initiation. Preliminary results suggest that FGF-23 independently predicts cardiovascular death and incident heart failure, but not acute myocardial infarction or stroke.
In aggregate, these large prospective studies suggest that high FGF-23 levels may be associated with incident heart failure and cardiovascular death, rather than with incident coronary events, in individuals without overt CKD. As discussed below, these findings are in line with cross-sectional data from our HOM SWEET HOMe study, which is so far the largest non-CKD cohort in which the impact of FGF-23 on prevalent and incident cardiovascular disease is analysed [71].
FGF-23: marker or mediator of cardiovascular outcome?
The consistent association of hyperphosphatoninism with patient mortality across the spectrum of CKD, as well as with heart failure and (cardiovascular) mortality in several non-CKD patient cohorts, raises the question whether FGF-23 merely represents a ‘passive’ risk marker, or actively mediates cardiovascular damage.
In the former case, elevated FGF-23 levels might be an ‘innocent bystander’ that reflects the adverse cardiovascular implications of impaired phosphate metabolism and/or renal damage. Oral phosphate intake and hyperphosphataemia are considered major inducers of FGF-23 expression, and experimental data demonstrate that phosphate actively contributes to accelerated atherogenesis [72], arterial calcification [73] and left ventricular hypertrophy (LVH) [74]. Even though the prognostic impact of FGF-23 on cardiovascular events and/or mortality persisted after adjustment for serum phosphate in virtually all clinical trials, residual confounding cannot be ruled out: unlike FGF-23, serum phosphate measurements have a large biological variability [75]. Therefore, FGF-23 has been suggested to reflect serum phosphate levels over a longer period of time, similar to glycated haemoglobin in glucose homeostasis [76]. Thus, FGF-23 would be a more reliable indicator of elevated phosphate burden than a single phosphate measurement in epidemiological analyses.
In addition, as FGF-23 levels increase very early in the course of CKD, associations between elevated FGF-23 and adverse outcome in subjects without overt kidney disease might be caused by the well-known cardiovascular implications of even mild CKD. Creatinine-based estimations of GFR, which are used in most epidemiological studies for assessment of kidney function, do not always precisely detect early CKD. Moreover, all prospective studies that analysed hyperphosphatoninism in subjects without overt CKD did not specifically exclude subjects with eGFR <60 mL/min/1.73 m2. Despite adjustment for eGFR in multivariate analyses, residual confounding may occur because of limited precision of GFR estimation.
Alternatively, FGF-23 might actively induce cardiovascular damage, either via indirect or via direct pathways. Indirect pathways might imply hypovitaminosis D, as FGF-23 inhibits 1α-hydroxylase and thus lowers serum calcitriol levels, which is increasingly acknowledged as a non-traditional cardiovascular risk factor in subjects in CKD and non-CKD subjects.
The identification of direct pathophysiological pathways between hyperphosphatoninism and incident cardiovascular disease was more challenging, and our traditional understanding of FGF-23 biology seemingly precluded the existence of direct pathways: FGF-23 was deemed to require membrane-bound Klotho for exerting its endocrine effects at cellular level, and Klotho expression was considered to be restricted to traditional target organs of phosphate regulating hormones, namely to kidneys and parathyroid glands.
This traditional understanding was very recently refuted by Faul [77], who unveiled a Klotho-independent, causal role for FGF-23 in the pathogenesis of left ventricular hypertrophy (LVH).
Cardiovascular implications of hyperphosphatoninism
Inspired by cross-sectional echocardiographic studies which associated higher serum FGF-23 levels with prevalent LVH in CKD subjects and healthy subjects [78, 79], Faul et al. [80] set out to identify a direct cardiotoxic effect of FGF-23.
In a series of very elegant experiments, the authors first reported cell culture data which showed FGF-23 to directly cause pathological hypertrophic growth of isolated rat cardiomyocytes via FGF receptor-dependent activation of the calcineurin-NFAT (Nuclear factor of activated T-cells) signalling pathway, although cardiomyocytes did not express Klotho. Secondly, the authors replicated this pro-hypertrophic effect of FGF-23 on myocardial cells in vivo: in rodent models, intramyocardial as well as intravenous injection of FGF-23 induced pathological LVH. Similarly, LVH developed in Klotho-deficient mice, which constitutively overexpress FGF-23 in a futile attempt to combat hyperphosphataemia and hypervitaminosis D (which arise from Klotho deficiency and subsequent renal resistance to FGF-23 stimulation). Thirdly, the authors underscore the therapeutic potential of their findings in a rodent model of CKD, as administration of an unselective FGF-receptor blocker allowed the amelioration of the development of LVH in rats after 5/6 nephrectomy.
Finally, the experimental work was supplemented with a large set of serial echocardiographic studies from CRIC study participants. Expanding earlier findings from cross-sectional studies [78, 79], hyperphosphatoninism was associated with future LVH development in CKD patients who had normal ventricular geometry at study initiation.
LVH predisposes patients to subsequent development of left ventricular dysfunction and congestive heart failure [81, 82]. While experimental data on a direct pathophysiological contribution of hyperphosphatoninism in the sequential development of congestive heart failure are eagerly awaited, first large-scale clinical evidence for an association between elevated FGF-23 and impaired left ventricular systolic function came from our prospective HOM SWEET HOMe study.
In this study, we measured FGF-23 levels in 1309 patients who underwent elective coronary angiography between May 2007 and June 2010. Left ventricular function was assessed with ventriculography in 1022 out of 1309 patients. Cross-sectional data from all patients recruited before January 2010 were published recently [71] and up-dated for the present review by adding data from patients recruited between February and June 2010. In line with our initial report, HOM SWEET HOMe patients with impaired left ventricular function had higher FGF-23 levels; the same held true after exclusion of patients with an eGFR of <60 mL/min/1.73 m2 [71]. Notably, these findings are in line with supplementary information reported from the Heart and Soul Study, in which baseline ejection fraction was similarly correlated with serum FGF-23 levels.
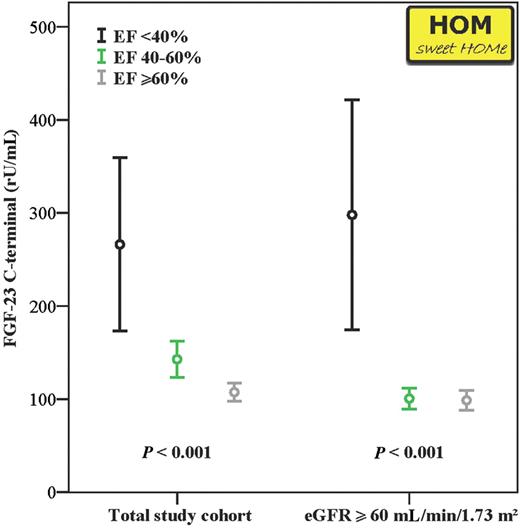
FGF-23 levels among 1309 HOM SWEET HOMe participants after stratifying for ejection fraction (EF; categorized as 40, 40–59 and ≥60%) in the total study cohort (left columns), and in individuals with intact renal function (eGFR ≥60 mL/min/1.73 m2; right columns). Data are presented as mean + SEM.
We additionally re-analysed the association of hyperphosphatoninism with atrial fibrillation among all HOM SWEET HOMe participants. In line with our earlier report, higher FGF-23 levels were found in patients with prevalent atrial fibrillation before and after exclusion of patients with an eGFR of <60 mL/min/1.73 m2, again pointing towards a relevant conjunction between hyperphosphatoninism and cardiac damage (Figure 3).
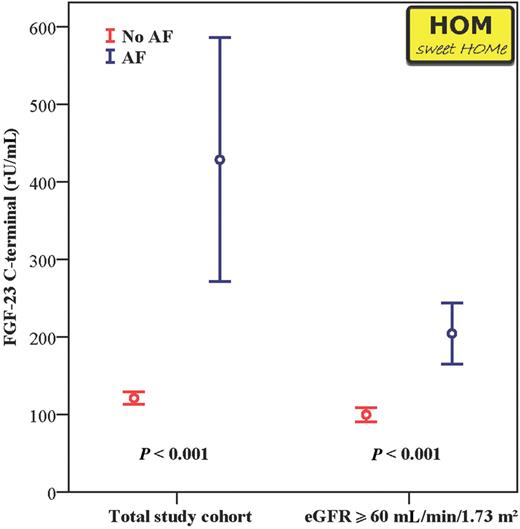
FGF-23 levels among 1309 HOM SWEET HOMe participants after stratifying for the presence of atrial fibrillation (AF) in the total study cohort (left columns), and in individuals with intact renal function (eGFR ≥60 mL/min/1.73 m2; right columns). Data are presented as mean + SEM.
Notably, HOM SWEET HOMe did not find a correlation between serum FGF-23 levels and presence or severity of coronary artery disease. At a first glance, this finding may appear somewhat counterintuitive, given the strong association between FGF-23 and serum phosphate levels, and the large experimental and clinical evidence for a significant contribution of phosphate to cardiovascular disease [83]. Nevertheless, these findings are in line with cross-sectional studies in patients without overt CKD, which did not find a rise in serum FGF-23 levels among patients with coronary artery stenosis at coronary multislice computed tomography [84] or with the presence of arterial stenosis at whole-body magnetic resonance angiography [85] after adjustment for confounders.
Will FGF-23 become a novel therapeutic target in cardiovascular medicine?
Integrating recent pivotal experimental data on a Klotho-independent cardiotoxic impact of elevated FGF-23 into topical epidemiological evidence for a detrimental effect of hyperphosphatoninism, the following pathophysiological concept might be hypothesized: diet and/or CKD-associated chronic phosphate loading stimulates osteoblastic FGF-23 secretion. The resulting hyperphosphatoninism, which physiologically aims to keep serum phosphate levels within seemingly normal ranges, activates cardiomyocytes, which stepwise induces the development of LVH and subsequently of left ventricular dysfunction and congestive heart failure. While presently neither experimental nor clinical data support a direct contribution of FGF-23 in accelerated atherogenesis, phosphate loading may actively propagate vascular calcification and atherosclerosis. This may induce further myocardial injury both by increasing arterial stiffness and subsequently cardiac afterload and by hindering coronary blood supply of cardiomyocytes (compare Figure 4).
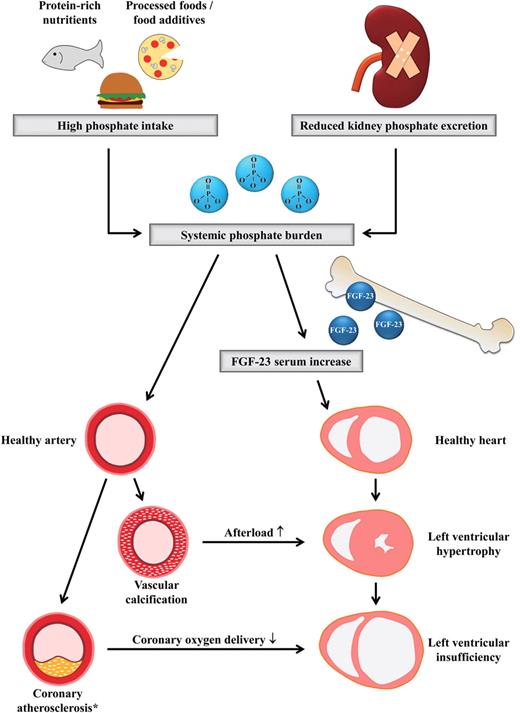
Simplified hypothetical patholophysiological concept which integrates the contribution of systemic phosphate burden and hyperphosphatoninism in the development of cardiovascular disease. Details are discussed in the text. *The selective contribution of serum phosphate to atherosclerotic plaque development is yet under discussion.
Admittedly, substantially more evidence from future clinical and experimental studies is needed to further support this hypothetical concept. Nevertheless, the persisting high cardiovascular death toll in CKD patients and the poor evidence for any beneficial intervention mandate us to conceive novel treatment strategies in CKD and to test their efficacy in clinical trials.
Three distinct therapeutic strategies for hyperphosphatoninism have been suggested: oral phosphate binders, FGF-23-blocking agents and FGF-R antagonists. Only oral phosphate binders are presently available. Their capacity to affect serum FGF-23 levels has been clearly demonstrated [86–88], and a blueprint for a large-scale, endpoint-driven clinical trial has been suggested [45]. Moreover, while oral phosphate binders are less specific agents than FGF-23-blocking agents or FGF-R antagonists, they bear the advantage of tackling two components of CKD-MBD by lowering both systemic phosphate burden and serum FGF-23. In theory, such a dual treatment strategy should allow broader vasculo- and cardioprotection.
Although FGF-23-blocking agents and FGF-R antagonists may be welcomed as modern and disease-specific drugs in hyperphosphatoninism, they bear the risk of elevating serum phosphate levels and systemic phosphate burden by inhibiting renal phosphate excretion. Of note, a better understanding of organ-specific FGF-R expression may allow to develop selective blockers of cardiac FGF-R, thus inhibiting hyperphosphatoninism-induced LVH, while leaving renal phosphate excretion unaffected.
In summary, hyperphosphatoninism has been acknowledged as a central component of CKD-MBD. A significant contribution of hyperphosphatoninism to cardiovascular disease has been suggested in large epidemiological studies and underscored by recent experimental data. Therapeutic interventions in hyperphosphatoninism should be investigated in adequately designed randomized trials.
Conflict of interest statement. S.S. has nothing to disclose. G.H.H. has been speaker for Shire. D.F. has been speaker and/or part of the advisory board for Shire, Amgen, Genzyme and FMC.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments